Note
This page was generated from
3_ligand_receptor_specfic_enrichment_analysis.ipynb.
Interactive online version:
![]() .
Some tutorial content may look better in light mode.
.
Some tutorial content may look better in light mode.
Ligand receptor specfic enrichment analysis.#
This notebook demonstrates ligand-specific enrichment analysis. Similar to the differential gene analysis, We can observe that the ligand receptor pair specifically enriched in a certain spatial region. This is done in the following three steps.
Construct a new anndata object, in which the columns of the matrix are cell-cell pairs, while the rows ligand-receptor mechanisms.
Based on above matrix, do dimensionality reduction,spatial clustering and differential mechanisms (ligand-receptor) analysis, to find the specifically enriched in a certain spatial region.
Reference: Micha Sam Brickman Raredon, Junchen Yang, Neeharika Kothapalli, Naftali Kaminski, Laura E. Niklason,Yuval Kluger. Comprehensive visualization of cell-cell interactions in single-cell and spatial transcriptomics with NICHES. doi: https://doi.org/10.1101/2022.01.23.477401
[ ]:
import os
import spateo as st
import dynamo as dyn
import matplotlib.pyplot as plt
import seaborn as sns
import pandas as pd
import numpy as np
Load data#
We will be using a axolotl dataset from [Wei et al., 2022] (https://doi.org/10.1126/science.abp9444).
Here, we can get data directly from the functionst.sample.axolotl or link:
axolotl_2DPI: https://www.dropbox.com/s/7w2jxf41xazrqxo/axolotl_2DPI.h5ad?dl=1
[3]:
adata = st.sample_data.axolotl(filename='axolotl_2DPI.h5ad')
adata
[3]:
AnnData object with n_obs × n_vars = 7668 × 27324
obs: 'CellID', 'spatial_leiden_e30_s8', 'Batch', 'cell_id', 'seurat_clusters', 'inj_uninj', 'D_V', 'inj_M_L', 'Annotation'
var: 'Axolotl_ID', 'hs_gene'
uns: 'Annotation_colors', '__type', 'color_key'
obsm: 'X_spatial', 'spatial'
layers: 'counts', 'log1p'
obsp: 'connectivities', 'distances', 'spatial_connectivities', 'spatial_distances'
[4]:
adata.var['new_name'] = adata.var.index
adata.var.index = adata.var['Axolotl_ID']
[4]:
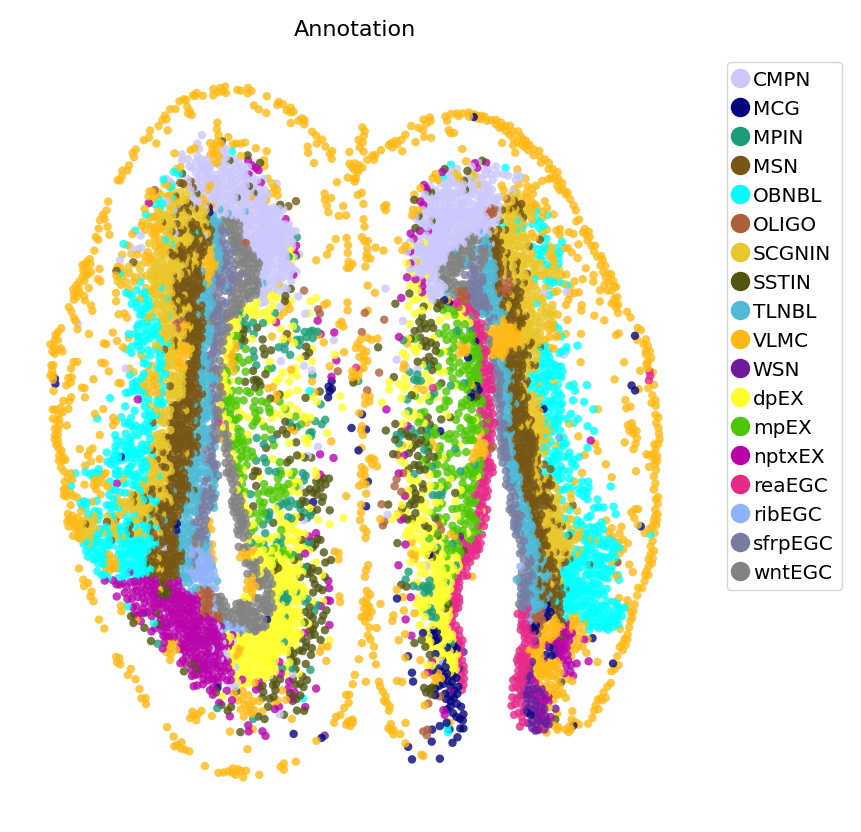
st.pl.space(adata,
color=['Annotation'],
pointsize=0.2,
color_key=adata.uns['color_key'],
show_legend='upper left',
figsize=(5, 5))
Construct new adata.#
First, calculate spatial nearest neighbor graph, limiting the nearest neighbor per cell to k. This function returns another anndata object, in which the columns of the matrix are cell-cell pairs, while the rows ligand-receptor mechanisms.
[5]:
weights_graph, distance_graph, adata = st.tl.weighted_spatial_graph(
adata,
n_neighbors=10,
)
|-----> <insert> spatial_connectivities to obsp in AnnData Object.
|-----> <insert> spatial_distances to obsp in AnnData Object.
|-----> <insert> spatial_neighbors to uns in AnnData Object.
|-----> <insert> spatial_neighbors.indices to uns in AnnData Object.
|-----> <insert> spatial_neighbors.params to uns in AnnData Object.
|-----> <insert> spatial_weights to obsp in AnnData Object.
[6]:
adata_n2n = st.tl.niches(adata,
path='/DATA/User/zuolulu/spateo-release/spateo/tools/database/',
layer=None,
species='axolotl',
system='niches_n2n',
method='sum')
adata_n2n
[6]:
AnnData object with n_obs × n_vars = 7668 × 916
obs: 'cell_pair_name'
Copy some of the other attributes from adata to adata_n2n. (Note assignment based on actual data).
[10]:
adata_n2n.uns['__type'] = 'UMI'
adata_n2n.obsm['spatial'] = adata.obsm['spatial']
Downstream analysis.#
Then, we can do dimensionality reduction , spatial clustering and differential mechanisms (ligand-receptor) analysis based on this new object. Therefore, we can find the ligand-receptor pairs which are specially enriched in a certain region.
preprocessing
[11]:
adata_n2n.obs['n_counts'] = adata_n2n.X.sum(axis=1).A1
adata_n2n.uns["pp"] = {}
adata_n2n.var_names_make_unique()
dyn.pl.basic_stats(adata_n2n)
adata_n2n.layers['raw'] = adata_n2n.X
dyn.pp.normalize_cell_expr_by_size_factors(adata_n2n, layers="X")
adata_n2n.layers['norm_log1p'] = adata_n2n.X.copy()
adata_n2n.X = adata_n2n.layers['raw'].copy()
st.tl.pearson_residuals(adata_n2n, n_top_genes=None)
adata_n2n
|-----> rounding expression data of layer: X during size factor calculation
|-----> size factor normalize following layers: ['X']
|-----> applying <ufunc 'log1p'> to layer<X>
|-----> set adata <X> to normalized data.
|-----> <insert> pp.norm_method to uns in AnnData Object.
[11]:
AnnData object with n_obs × n_vars = 7668 × 916
obs: 'cell_pair_name', 'n_counts', 'nGenes', 'nCounts', 'pMito', 'Size_Factor', 'initial_cell_size'
var: 'nCells', 'nCounts'
uns: '__type', 'pp'
obsm: 'spatial', 'pearson_residuals'
layers: 'raw', 'norm_log1p'
[12]:
bad_genes = np.isnan(adata_n2n.obsm["pearson_residuals"].sum(0))
bad_genes.sum()
st.tl.pca_spateo(adata=adata_n2n, X_data=adata_n2n.obsm["pearson_residuals"]
[:, ~bad_genes], n_pca_components=30, pca_key="X_pca", random_state=1)
dyn.tl.neighbors(adata_n2n, n_neighbors=30)
|-----> Runing PCA on user provided data...
|-----> Start computing neighbor graph...
|-----------> X_data is None, fetching or recomputing...
|-----> fetching X data from layer:None, basis:pca
|-----> method arg is None, choosing methods automatically...
|-----------> method ball_tree selected
|-----> <insert> connectivities to obsp in AnnData Object.
|-----> <insert> distances to obsp in AnnData Object.
|-----> <insert> neighbors to uns in AnnData Object.
|-----> <insert> neighbors.indices to uns in AnnData Object.
|-----> <insert> neighbors.params to uns in AnnData Object.
[12]:
AnnData object with n_obs × n_vars = 7668 × 916
obs: 'cell_pair_name', 'n_counts', 'nGenes', 'nCounts', 'pMito', 'Size_Factor', 'initial_cell_size'
var: 'nCells', 'nCounts'
uns: '__type', 'pp', 'neighbors'
obsm: 'spatial', 'pearson_residuals', 'X_pca'
layers: 'raw', 'norm_log1p'
obsp: 'distances', 'connectivities'
clustering
[13]:
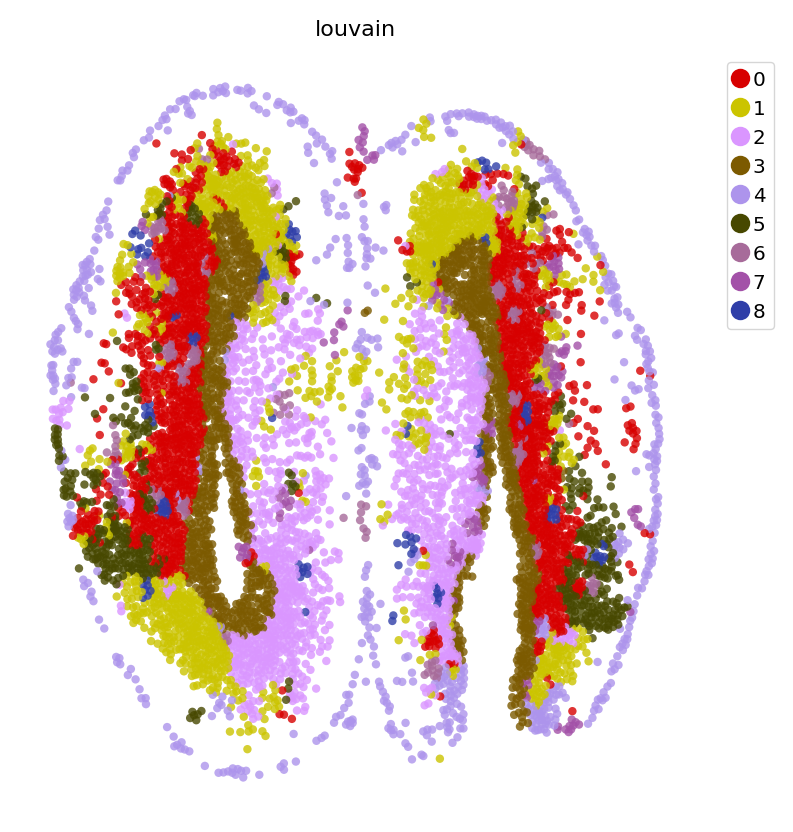
dyn.tl.louvain(adata_n2n, resolution=2)
st.pl.space(adata_n2n,
color=['louvain'],
pointsize=0.2,
figsize=(5, 5),
show_legend="upper left")
dyn.tl.reduceDimension(adata_n2n)
st.pl.space(adata_n2n,
color=['louvain'],
figsize=(5, 5),
space="umap",
pointsize=0.2)
|-----> accessing adj_matrix_key=distances built from args for clustering...
|-----> Detecting communities on graph...
|-----------> Converting graph_sparse_matrix to networkx object
|-----> [Community clustering with louvain] in progress: 100.0000%
|-----> [Community clustering with louvain] finished [412.6376s]
|-----> retrive data for non-linear dimension reduction...
|-----> perform umap...
|-----> [dimension_reduction projection] in progress: 100.0000%
|-----> [dimension_reduction projection] finished [43.6265s]

differential analysis
[42]:
# (optional: just Suitable for axolotl, homologous gene to human)
#adata_n2n.var['lr_pair_name'] = adata_n2n.var.index
df = adata_n2n.var['lr_pair_name'].str.split('-', expand=True)
df.columns = ['ligand', 'receptor']
##
df1 = adata.var
df1.index.name = None
df2 = pd.merge(df, df1, left_on='ligand', right_on='Axolotl_ID').drop(
['Axolotl_ID', 'new_name'], axis=1)
df2.columns = ['ligand', 'receptor', 'ligand_human']
df3 = pd.merge(df2, df1, left_on='receptor',
right_on='Axolotl_ID').drop(['Axolotl_ID', 'new_name'], axis=1)
df3.columns = ['ligand', 'receptor', 'ligand_human', 'receptor_human']
df3['ligand_human'] = df3['ligand_human'].astype('str')
df3['receptor_human'] = df3['receptor_human'].astype('str')
df3['lr_pair'] = df3["ligand_human"] + "-" + df3["receptor_human"]
adata_n2n.var['lr_pair'] = df3['lr_pair'].tolist()
adata_n2n.var.index = adata_n2n.var['lr_pair']
[33]:
adata_n2c_marker = st.tl.find_all_cluster_degs(
adata_n2n, group='louvain', genes=None, n_jobs=1)
identifying top markers for each group: 916it [00:04, 223.56it/s]
identifying top markers for each group: 916it [00:04, 214.35it/s]
identifying top markers for each group: 916it [00:04, 199.74it/s]
identifying top markers for each group: 916it [00:05, 178.17it/s]
identifying top markers for each group: 916it [00:04, 224.85it/s]
identifying top markers for each group: 916it [00:03, 236.41it/s]
identifying top markers for each group: 916it [00:04, 217.55it/s]
identifying top markers for each group: 916it [00:04, 201.59it/s]
identifying top markers for each group: 916it [00:04, 204.19it/s]
[34]:
# top_n_markers
marker_genes_dict = st.tl.top_n_degs(
adata_n2c_marker, group='louvain', top_n_genes=3)
[35]:
deg_table = st.tl.top_n_degs(adata_n2c_marker, group='louvain',
only_deg_list=False, sort_by='cosine_score', top_n_genes=3)
[36]:
markers = deg_table['gene'].unique().tolist()
adata_n2n.var_names_make_unique()
adata_n2n.obs['louvain'] = adata_n2n.obs['louvain'].astype('category')
[37]:
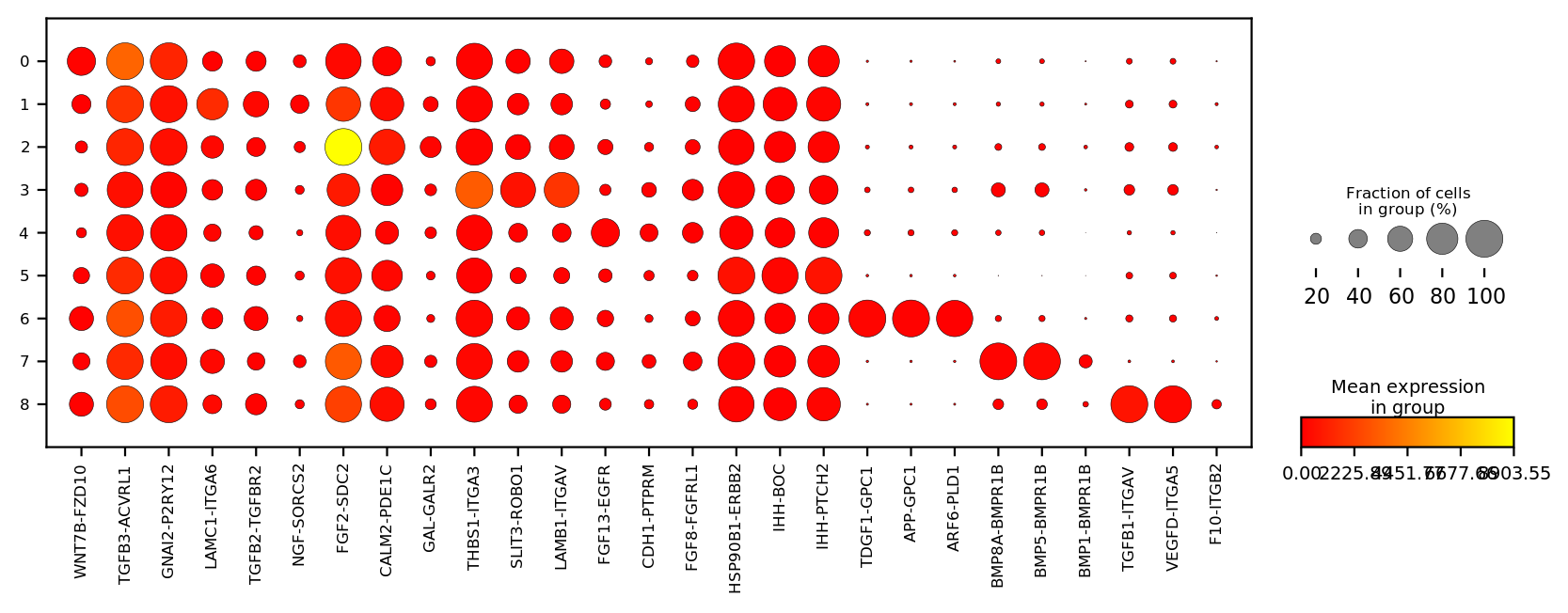
st.pl.dotplot(adata_n2n,
var_names=markers,
cat_key='louvain',
cmap='autumn',
figsize=(10, 3),
save_show_or_return='return')
[37]:
(<Figure size 1000x300 with 4 Axes>,
{'mainplot_ax': <AxesSubplot:>,
'size_legend_ax': <AxesSubplot:title={'center':'Fraction of cells\nin group (%)'}>,
'color_legend_ax': <AxesSubplot:title={'center':'Mean expression\nin group'}>})
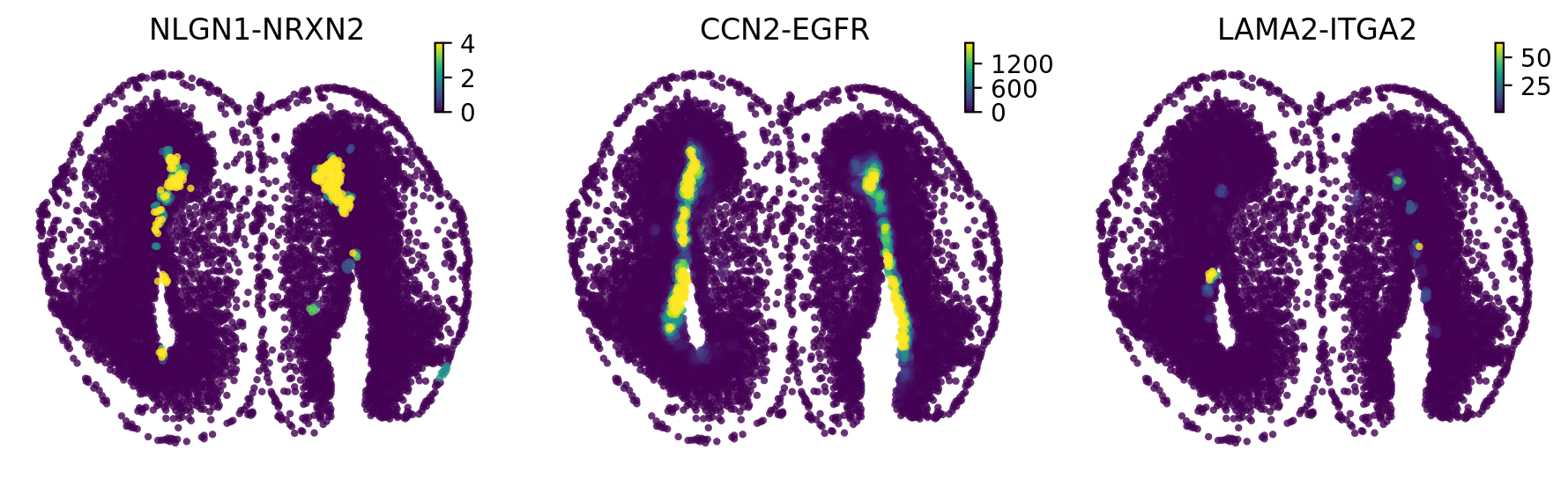
Plot spatial in situ expression map
[41]:
st.pl.space(adata_n2n,
color=marker_genes_dict['3'],
pointsize=0.2,
ncols=3,
figsize=(4, 3),
show_legend="upper left")