Note
This page was generated from
3_morphogenesis_feature.ipynb.
Interactive online version:
![]() .
Some tutorial content may look better in light mode.
.
Some tutorial content may look better in light mode.
3.morphogenesis features¶
Packages¶
[1]:
import os
import sys
import gseapy as gp
import numpy as np
import dynamo as dyn
import spateo as st
/home/yao/anaconda3/envs/BGIpy38_tf2/lib/python3.8/site-packages/requests/__init__.py:102: RequestsDependencyWarning: urllib3 (1.26.9) or chardet (5.0.0)/charset_normalizer (2.0.12) doesn't match a supported version!
warnings.warn("urllib3 ({}) or chardet ({})/charset_normalizer ({}) doesn't match a supported "
|-----> setting visualization default mode in dynamo. Your customized matplotlib settings might be overritten.
/home/yao/.local/lib/python3.8/site-packages/nxviz/__init__.py:18: UserWarning:
nxviz has a new API! Version 0.7.3 onwards, the old class-based API is being
deprecated in favour of a new API focused on advancing a grammar of network
graphics. If your plotting code depends on the old API, please consider
pinning nxviz at version 0.7.3, as the new API will break your old code.
To check out the new API, please head over to the docs at
https://ericmjl.github.io/nxviz/ to learn more. We hope you enjoy using it!
(This deprecation message will go away in version 1.0.)
warnings.warn(
/home/yao/anaconda3/envs/BGIpy38_tf2/lib/python3.8/site-packages/spaghetti/network.py:36: FutureWarning:
The next major release of pysal/spaghetti (2.0.0) will drop support for all ``libpysal.cg`` geometries. This change is a first step in refactoring ``spaghetti`` that is expected to result in dramatically reduced runtimes for network instantiation and operations. Users currently requiring network and point pattern input as ``libpysal.cg`` geometries should prepare for this simply by converting to ``shapely`` geometries.
Data source¶
[2]:
# The anndata object is reconstructed by ``2.morphogenesis vector fields and trajectories``
stage_adata = st.sample_data.drosophila(filename="E7-9h_cellbin_tdr_v3_midgut.h5ad")
stage_adata = stage_adata[:, np.sum(stage_adata.layers["counts_X"], axis=0) != 0]
stage_adata.X = stage_adata.layers["counts_X"].copy()
stage_adata.uns["pp"] = {}
dyn.pp.normalize_cell_expr_by_size_factors(adata=stage_adata, layers="X", X_total_layers=True, skip_log=True)
# The pc model and mesh model are reconstructed by ``2.morphogenesis vector fields and trajectories``
stage_pc = st.tdr.read_model(f"vector_pc_model_midgut.vtk")
stage_mesh = st.tdr.read_model(f"vector_mesh_model_midgut.vtk")
cpo = [(531.4285063139628 / 1.5, 1120.6071331378873 / 1.5, 226.6879332984771 / 1.5),
(1.9670869138005287, -6.902875264241757, -2.2120172004343885),
(-0.1349909265808914, -0.13586573883518835, 0.9814876212931068)]
stage_adata
|-----> rounding expression data of layer: X during size factor calculation
|-----> size factor normalize following layers: ['X']
|-----? `total_szfactor` is not `None` and it is not in adata object.
|-----> skipping log transformation as input requires...
|-----> applying None to layer<X>
|-----> set adata <X> to normalized data.
|-----> <insert> pp.norm_method to uns in AnnData Object.
[2]:
AnnData object with n_obs × n_vars = 2326 × 7685
obs: 'area', 'slices', 'nGenes', 'nCounts', 'pMito', 'nMito', 'pass_basic_filter', 'scc', 'auto_anno', 'anno_cell_type', 'anno_tissue', 'anno_germ_layer', 'actual_stage', 'Size_Factor', 'initial_cell_size'
uns: 'PCs', 'VecFld_morpho', '__type', 'auto_anno_result', 'dendrogram_anno_cell_type', 'dendrogram_anno_germ_layer', 'dendrogram_anno_tissue', 'explained_variance_ratio_', 'fate_morpho', 'latter_models_align', 'neighbors', 'pca_mean', 'pca_valid_ind', 'pearson_X_neighbors', 'pp', 'rank_genes_groups', 'rank_genes_groups_anno_cell_type', 'rank_genes_groups_anno_germ_layer', 'rank_genes_groups_anno_tissue', 'scc', 'spatial'
obsm: '3d_align_spatial', 'V_cells_mapping', 'X_cells_mapping', 'align_spatial', 'bbox', 'before_3d_align_spatial', 'contour', 'pearson_X_pca', 'pearson_X_umap', 'spatial', 'tdr_spatial'
layers: 'counts_X', 'log1p_X', 'pearson_X', 'spliced', 'unspliced'
Generate the trajectory model¶
[3]:
trajectory_model = st.tdr.construct_trajectory(
adata=stage_adata,
fate_key="fate_morpho",
key_added="obs_index",
n_sampling=500,
sampling_method="trn",
label=np.asarray(stage_adata.obs.index),
)
|-----> [Running TRN] in progress: 100.0000%
|-----> [Running TRN] finished [23.7339s]
Morphogenesis features¶
[5]:
glm_dict = {}
Velocity¶
[6]:
key = "velocity"
st.tdr.morphofield_velocity(adata=stage_adata, vf_key="VecFld_morpho", key_added=key,)
stage_adata.obsm["velocity"]
[6]:
array([[-1.91148383e-03, -1.82671905e-03, -2.71598635e-03],
[-1.95678207e-03, -1.91036185e-03, -2.87477111e-03],
[-1.55106979e-03, -1.68945969e-03, -2.41216961e-03],
...,
[-2.10651796e-06, 1.09092371e-04, 1.71498103e-03],
[ 1.89100976e-06, 8.49589979e-05, 1.72119623e-03],
[-8.09060396e-05, 1.64108170e-04, 1.67981541e-03]])
Acceleration¶
[7]:
key = "acceleration"
glm_key = f"glm_degs_{key}"
st.tdr.morphofield_acceleration(adata=stage_adata, vf_key="VecFld_morpho", key_added=key,)
st.tl.glm_degs(adata=stage_adata, layer=None, fullModelFormulaStr=f'~cr({key}, df=3)', key_added=glm_key, qval_threshold=0.01, llf_threshold=-2500)
glm_data = stage_adata.uns[glm_key]["glm_result"]
glm_dict[key] = glm_data
glm_data
|-----> [Calculating acceleration] in progress: 100.0000%
|-----> [Calculating acceleration] finished [0.0412s]
|-----? Gene expression matrix must be normalized by the size factor, please check if the input gene expression matrix is correct.If you don't have the size factor normalized gene expression matrix, please run `dynamo.pp.normalize_cell_expr_by_size_factors(skip_log = True)`.
|-----> [Detecting genes via Generalized Additive Models (GAMs)] in progress: 100.0000%
|-----> [Detecting genes via Generalized Additive Models (GAMs)] finished [375.0744s]
[7]:
| status | family | log-likelihood | pval | qval | |
|---|---|---|---|---|---|
| Bacc | ok | NB2 | -3671.859375 | 4.873121e-11 | 7.489986e-09 |
| 28SrRNA-Psi:CR45862 | ok | NB2 | -4358.506348 | 6.277172e-10 | 6.432008e-08 |
| Cys | ok | NB2 | -4643.826172 | 4.387060e-08 | 2.148794e-06 |
| 28SrRNA-Psi:CR45859 | ok | NB2 | -4632.636230 | 2.513353e-06 | 4.769165e-05 |
| eEF1beta | ok | NB2 | -3303.432861 | 1.365682e-05 | 1.904767e-04 |
| Nph | ok | NB2 | -3012.880371 | 3.456467e-05 | 3.855290e-04 |
| fax | ok | NB2 | -2740.145508 | 5.348858e-05 | 5.373330e-04 |
| His3.3A | ok | NB2 | -3064.583496 | 6.052202e-05 | 5.887490e-04 |
| Fib | ok | NB2 | -2566.735840 | 2.022182e-04 | 1.532591e-03 |
| awd | ok | NB2 | -3193.938477 | 6.848338e-04 | 3.859636e-03 |
| mt:lrRNA | ok | NB2 | -6083.094238 | 7.883165e-04 | 4.318041e-03 |
| tsr | ok | NB2 | -3093.676758 | 9.017585e-04 | 4.782618e-03 |
| 28SrRNA-Psi:CR40596 | ok | NB2 | -3915.597412 | 1.051448e-03 | 5.379748e-03 |
| pAbp | ok | NB2 | -3751.640137 | 1.692311e-03 | 7.820453e-03 |
[8]:
st.pl.glm_fit(
adata=stage_adata,
gene=glm_data.index.tolist(),
ncols=5,
feature_x=key,
feature_y="expression",
feature_fit="mu",
glm_key=glm_key,
lowess=False,
frac=0.2,
save_show_or_return="all",
)
st.pl.glm_fit(
adata=stage_adata,
gene=glm_data.index.tolist(),
ncols=5,
feature_x=key,
feature_y="expression",
feature_fit="mu",
glm_key=glm_key,
lowess=True,
frac=0.2,
save_show_or_return="all",
)
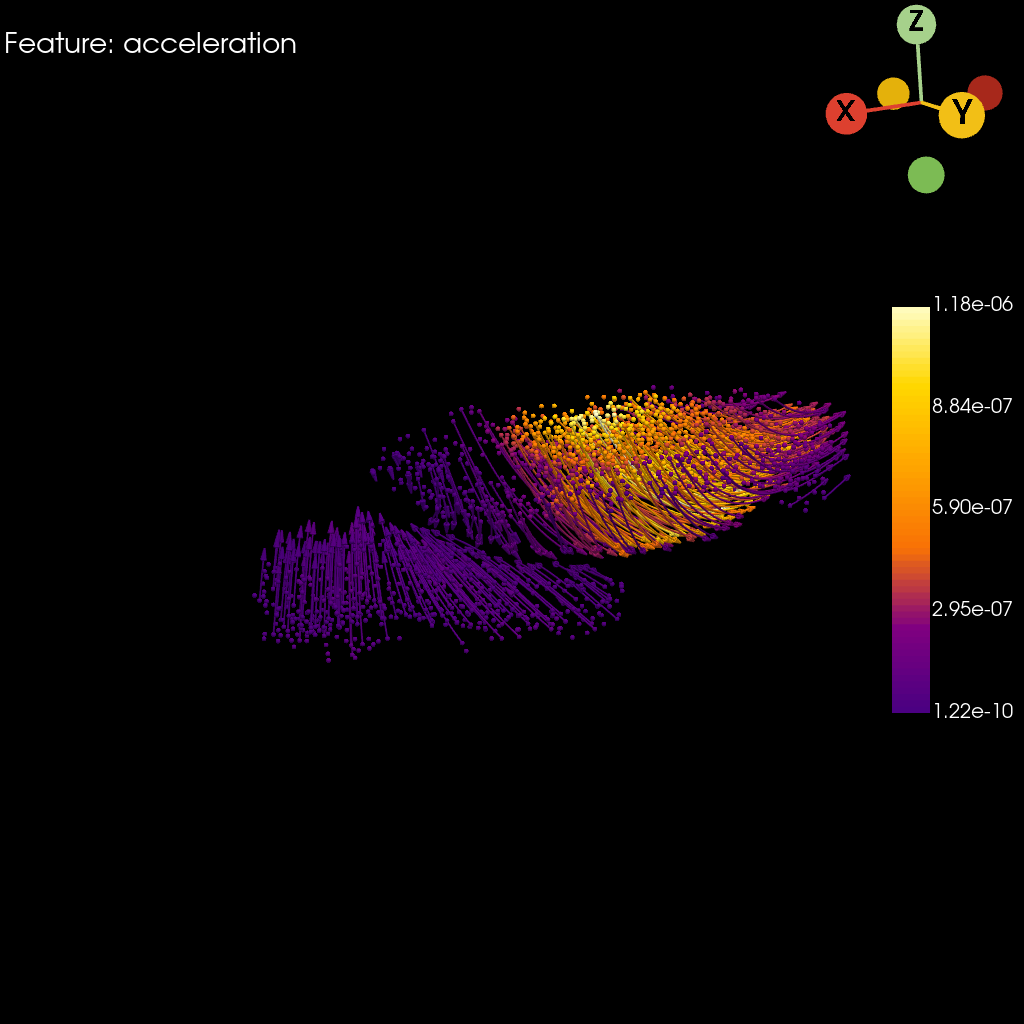
st.pl.acceleration(
adata=stage_adata,
model=st.tdr.collect_models([stage_pc, trajectory_model]),
acceleration_key=key,
colormap="default_cmap",
jupyter="static",
model_style=["points", "wireframe"],
model_size=[5, 2],
cpo=cpo,
window_size=(1024, 1024),
legend_kwargs=dict(title="", fmt="%.2e", legend_loc=(0.87, 0.3), label_font_size=20),
)
Saving figure to develop/develop_midgut/genesis_feature/E7-9h_midgut_acceleration/E7-9h_midgut_glm_degs_fit_acceleration.pdf...
Done

Saving figure to develop/develop_midgut/genesis_feature/E7-9h_midgut_acceleration/E7-9h_midgut_glm_degs_fit_acceleration_lowess.pdf...
Done

[9]:
st.tdr.add_model_labels(
model=trajectory_model,
key_added=key,
labels=np.asarray(stage_adata[np.asarray(trajectory_model.point_data["obs_index"])].obs[key]),
where="point_data",
inplace=True,
)
cells_linear_models = st.tdr.construct_genesis_X(
stages_X=[stage_adata.obsm["3d_align_spatial"], stage_adata.obsm["X_cells_mapping"]],
n_spacing=100-1,
key_added=key,
label=[np.asarray(stage_adata.obs[key])] * 100,
)
st.pl.three_d_animate(
models=cells_linear_models,
stable_model=trajectory_model,
key=key,
stable_kwargs=dict(
key=key,
model_style="wireframe",
model_size=2,
ambient=0.5,
opacity=0.3,
colormap="default_cmap",
show_legend=False,
),
filename=f"E7-9h_midgut_{key}_linear_movie_with_trajectory.mp4",
model_style="points",
model_size=5,
ambient=0.5,
colormap="default_cmap",
show_legend=True,
legend_kwargs=dict(title="", fmt="%.2e", legend_loc=(0.87, 0.3), label_font_size=20),
off_screen=True,
jupyter=False,
background="black",
cpo=cpo,
window_size=(1024, 1024),
)
st.pl.three_d_animate(
models=cells_linear_models,
stable_model=None,
key=key,
filename=f"E7-9h_midgut_{key}_linear_movie.mp4",
model_style="points",
model_size=5,
ambient=0.5,
colormap="default_cmap",
show_legend=True,
legend_kwargs=dict(title="", fmt="%.2e", legend_loc=(0.87, 0.3), label_font_size=20),
off_screen=True,
jupyter=False,
background="black",
cpo=cpo,
window_size=(1024, 1024),
)
Curvature¶
[10]:
key = "curvature"
glm_key = f"glm_degs_{key}"
st.tdr.morphofield_curvature(adata=stage_adata, vf_key="VecFld_morpho", key_added=key,)
st.tl.glm_degs(adata=stage_adata, layer=None, fullModelFormulaStr=f'~cr({key}, df=3)', key_added=glm_key, qval_threshold=0.01, llf_threshold=-2500)
glm_data = stage_adata.uns[glm_key]["glm_result"]
glm_dict[key] = glm_data
glm_data
|-----> [Calculating acceleration] in progress: 100.0000%
|-----> [Calculating acceleration] finished [0.0304s]
|-----> [Calculating curvature] in progress: 100.0000%
|-----> [Calculating curvature] finished [0.0381s]
|-----? Gene expression matrix must be normalized by the size factor, please check if the input gene expression matrix is correct.If you don't have the size factor normalized gene expression matrix, please run `dynamo.pp.normalize_cell_expr_by_size_factors(skip_log = True)`.
|-----> [Detecting genes via Generalized Additive Models (GAMs)] in progress: 100.0000%
|-----> [Detecting genes via Generalized Additive Models (GAMs)] finished [287.0102s]
[10]:
| status | family | log-likelihood | pval | qval | |
|---|---|---|---|---|---|
| Ance | ok | NB2 | -3339.842529 | 5.916981e-13 | 1.136800e-09 |
| Fib | ok | NB2 | -2560.309814 | 3.273910e-07 | 8.064102e-06 |
| 28SrRNA-Psi:CR40596 | ok | NB2 | -3911.105713 | 1.177748e-05 | 1.544538e-04 |
| mt:ND5 | ok | NB2 | -2526.593506 | 2.593650e-05 | 2.823258e-04 |
| RpL24-like | ok | NB2 | -3122.291748 | 2.842352e-05 | 3.025412e-04 |
| Cys | ok | NB2 | -4650.455078 | 3.319294e-05 | 3.410264e-04 |
| 28SrRNA-Psi:CR45862 | ok | NB2 | -4370.237793 | 7.809990e-05 | 6.751381e-04 |
| RNASEK | ok | NB2 | -2597.170898 | 1.023775e-04 | 8.238442e-04 |
| His2Av | ok | NB2 | -3063.656494 | 1.208075e-04 | 9.377835e-04 |
| sesB | ok | NB2 | -2753.985596 | 1.806184e-04 | 1.286111e-03 |
| COX7C | ok | NB2 | -2909.798096 | 3.349247e-04 | 2.114103e-03 |
| Obp99a | ok | NB2 | -3229.527588 | 4.188366e-04 | 2.513024e-03 |
| Cyp1 | ok | NB2 | -3645.378418 | 8.155234e-04 | 4.231801e-03 |
| chic | ok | NB2 | -2536.232910 | 8.432105e-04 | 4.340303e-03 |
| RpS16 | ok | NB2 | -4785.575684 | 1.652613e-03 | 7.349729e-03 |
[11]:
st.pl.glm_fit(
adata=stage_adata,
gene=glm_data.index.tolist(),
ncols=5,
feature_x=key,
feature_y="expression",
feature_fit="mu",
glm_key=glm_key,
lowess=False,
frac=0.2,
save_show_or_return="both",
)
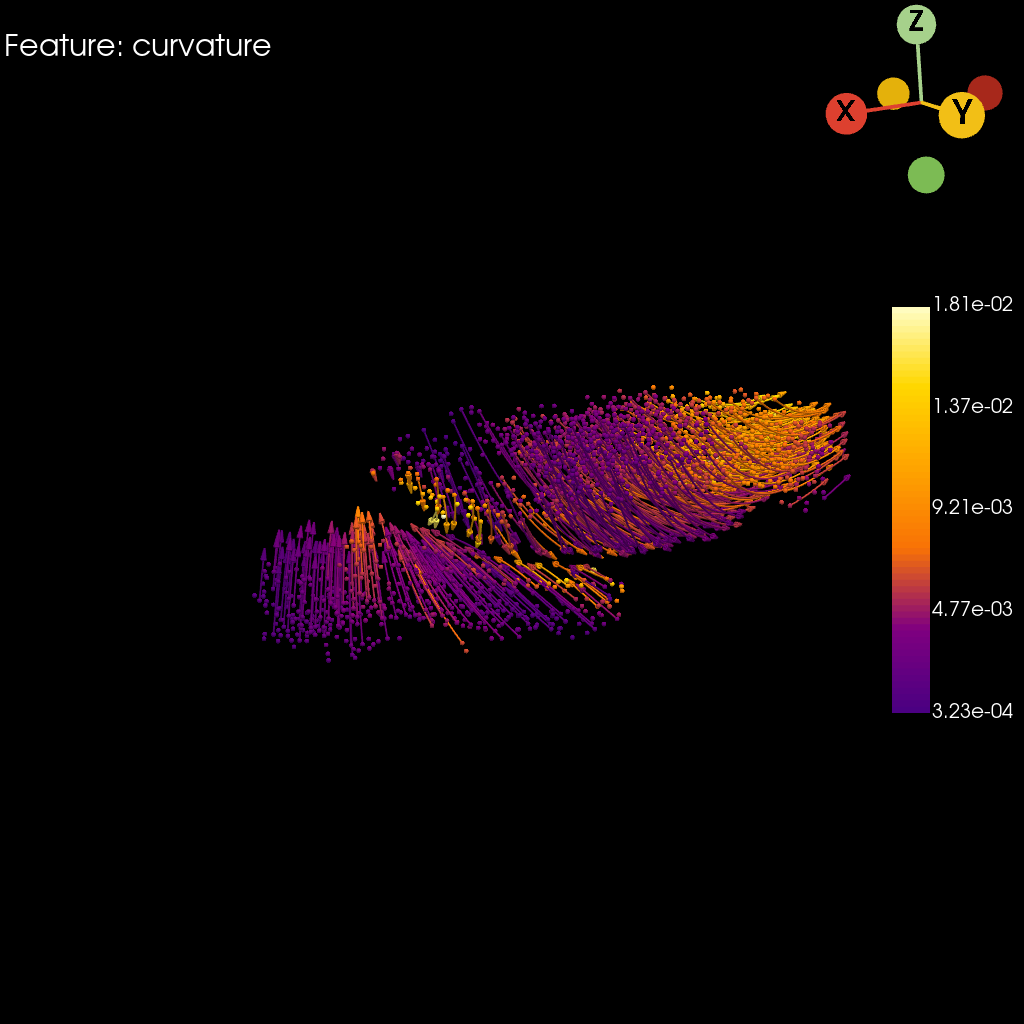
st.pl.curvature(
adata=stage_adata,
model=st.tdr.collect_models([stage_pc, trajectory_model]),
curvature_key=key,
colormap="default_cmap",
jupyter="static",
model_style=["points", "wireframe"],
model_size=[5, 2],
cpo=cpo,
window_size=(1024, 1024),
legend_kwargs=dict(title="", fmt="%.2e", legend_loc=(0.87, 0.3), label_font_size=20)
)
Saving figure to develop/develop_midgut/genesis_feature/E7-9h_midgut_curvature/E7-9h_midgut_glm_degs_fit_curvature.pdf...
Done

[12]:
st.tdr.add_model_labels(
model=trajectory_model,
key_added=key,
labels=np.asarray(stage_adata[np.asarray(trajectory_model.point_data["obs_index"])].obs[key]),
where="point_data",
inplace=True,
)
cells_linear_models = st.tdr.construct_genesis_X(
stages_X=[stage_adata.obsm["3d_align_spatial"], stage_adata.obsm["X_cells_mapping"]],
n_spacing=100-1,
key_added=key,
label=[np.asarray(stage_adata.obs[key])] * 100,
)
st.pl.three_d_animate(
models=cells_linear_models,
stable_model=trajectory_model,
key=key,
stable_kwargs=dict(
key=key,
model_style="wireframe",
model_size=2,
ambient=0.5,
opacity=0.3,
colormap="default_cmap",
show_legend=False,
),
filename=f"E7-9h_midgut_{key}_linear_movie_with_trajectory.mp4",
model_style="points",
model_size=5,
ambient=0.5,
colormap="default_cmap",
show_legend=True,
legend_kwargs=dict(title="", fmt="%.2e", legend_loc=(0.87, 0.3), label_font_size=20),
off_screen=True,
jupyter=False,
background="black",
cpo=cpo,
window_size=(1024, 1024),
)
st.pl.three_d_animate(
models=cells_linear_models,
stable_model=None,
key=key,
filename=f"E7-9h_midgut_{key}_linear_movie.mp4",
model_style="points",
model_size=5,
ambient=0.5,
colormap="default_cmap",
show_legend=True,
legend_kwargs=dict(title="", fmt="%.2e", legend_loc=(0.87, 0.3), label_font_size=20),
off_screen=True,
jupyter=False,
background="black",
cpo=cpo,
window_size=(1024, 1024),
)
Curl¶
[13]:
key = "curl"
glm_key = f"glm_degs_{key}"
st.tdr.morphofield_curl(adata=stage_adata, vf_key="VecFld_morpho", key_added=key,)
st.tl.glm_degs(adata=stage_adata, layer=None, fullModelFormulaStr=f'~cr({key}, df=3)', key_added=glm_key, qval_threshold=0.01, llf_threshold=-2500)
glm_data = stage_adata.uns[glm_key]["glm_result"]
glm_dict[key] = glm_data
glm_data
Calculating 3-D curl: 100%|██████████| 2326/2326 [00:00<00:00, 57530.08it/s]
|-----? Gene expression matrix must be normalized by the size factor, please check if the input gene expression matrix is correct.If you don't have the size factor normalized gene expression matrix, please run `dynamo.pp.normalize_cell_expr_by_size_factors(skip_log = True)`.
|-----> [Detecting genes via Generalized Additive Models (GAMs)] in progress: 100.0000%
|-----> [Detecting genes via Generalized Additive Models (GAMs)] finished [267.5932s]
[13]:
| status | family | log-likelihood | pval | qval | |
|---|---|---|---|---|---|
| Fib | ok | NB2 | -2560.906738 | 5.947219e-07 | 0.000015 |
| pAbp | ok | NB2 | -3744.795410 | 1.802494e-06 | 0.000038 |
| COX8 | ok | NB2 | -3233.309570 | 2.275150e-05 | 0.000267 |
| 28SrRNA-Psi:CR40596 | ok | NB2 | -3912.000488 | 2.882065e-05 | 0.000321 |
| Ance | ok | NB2 | -3358.986572 | 1.219921e-04 | 0.001011 |
| tsr | ok | NB2 | -3092.293213 | 2.260249e-04 | 0.001625 |
| Nph | ok | NB2 | -3015.566162 | 5.069769e-04 | 0.003041 |
| RpL24-like | ok | NB2 | -3125.715576 | 8.723374e-04 | 0.004005 |
| Bacc | ok | NB2 | -3688.385254 | 7.326030e-04 | 0.004059 |
| 28SrRNA-Psi:CR45859 | ok | NB2 | -4638.458984 | 8.491511e-04 | 0.004522 |
| fax | ok | NB2 | -2743.038330 | 9.650021e-04 | 0.005024 |
| NHP2 | ok | NB2 | -2605.645508 | 1.179141e-03 | 0.005976 |
| CNBP | ok | NB2 | -3570.270264 | 1.538176e-03 | 0.006671 |
| ATPsynG | ok | NB2 | -3090.621582 | 1.784211e-03 | 0.008645 |
| mt:ND1 | ok | NB2 | -3178.442871 | 2.424773e-03 | 0.009808 |
[14]:
st.pl.glm_fit(
adata=stage_adata,
gene=glm_data.index.tolist(),
ncols=5,
feature_x=key,
feature_y="expression",
feature_fit="mu",
glm_key=glm_key,
lowess=False,
frac=0.2,
save_show_or_return="both",
)
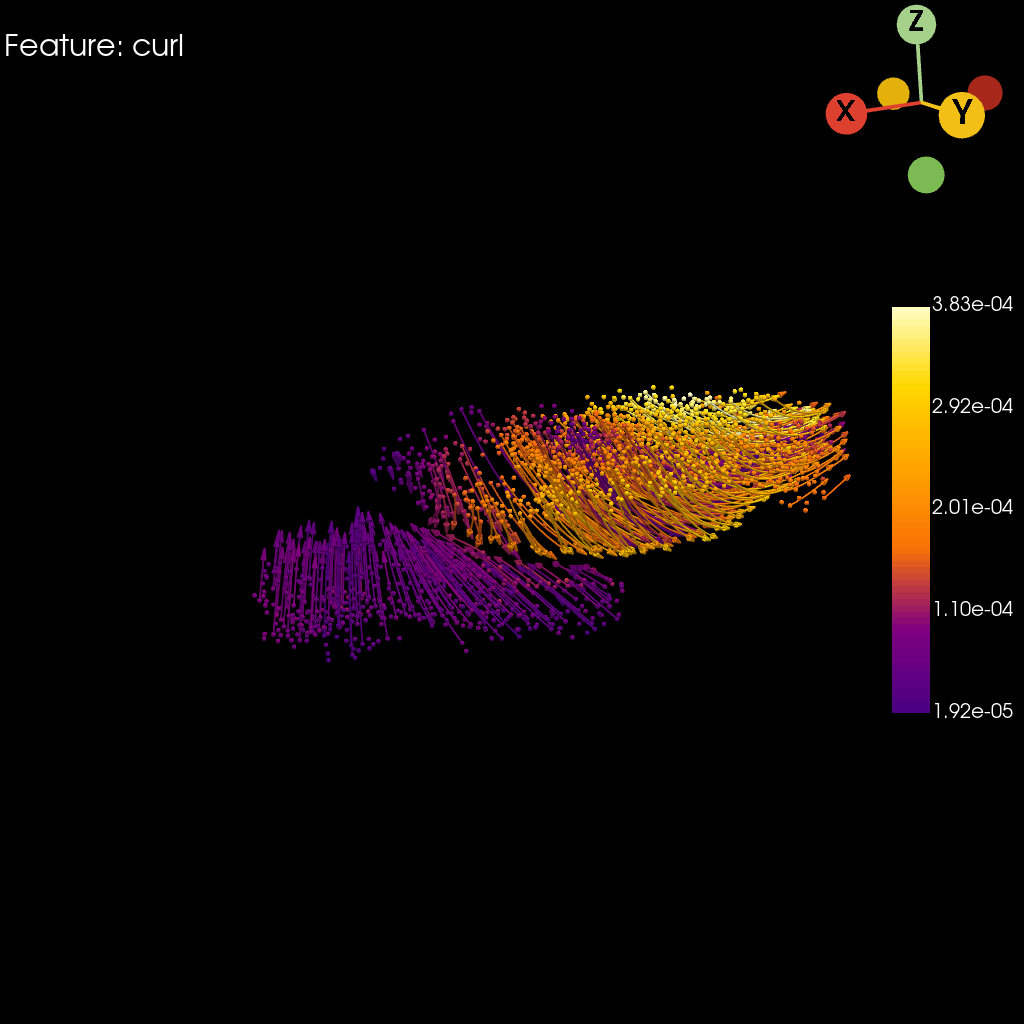
st.pl.curl(
adata=stage_adata,
model=st.tdr.collect_models([stage_pc, trajectory_model]),
curl_key=key,
colormap="default_cmap",
jupyter="static",
model_style=["points", "wireframe"],
model_size=[5, 2],
cpo=cpo,
window_size=(1024, 1024),
legend_kwargs=dict(title="", fmt="%.2e", legend_loc=(0.87, 0.3), label_font_size=20),
)
Saving figure to develop/develop_midgut/genesis_feature/E7-9h_midgut_curl/E7-9h_midgut_glm_degs_fit_curl.pdf...
Done

[15]:
st.tdr.add_model_labels(
model=trajectory_model,
key_added=key,
labels=np.asarray(stage_adata[np.asarray(trajectory_model.point_data["obs_index"])].obs[key]),
where="point_data",
inplace=True,
)
cells_linear_models = st.tdr.construct_genesis_X(
stages_X=[stage_adata.obsm["3d_align_spatial"], stage_adata.obsm["X_cells_mapping"]],
n_spacing=100-1,
key_added=key,
label=[np.asarray(stage_adata.obs[key])] * 100,
)
st.pl.three_d_animate(
models=cells_linear_models,
stable_model=trajectory_model,
key=key,
stable_kwargs=dict(
key=key,
model_style="wireframe",
model_size=2,
ambient=0.5,
opacity=0.3,
colormap="default_cmap",
show_legend=False,
),
filename=f"E7-9h_midgut_{key}_linear_movie_with_trajectory.mp4",
model_style="points",
model_size=5,
ambient=0.5,
colormap="default_cmap",
show_legend=True,
legend_kwargs=dict(title="", fmt="%.2e", legend_loc=(0.87, 0.3), label_font_size=20),
off_screen=True,
jupyter=False,
background="black",
cpo=cpo,
window_size=(1024, 1024),
)
st.pl.three_d_animate(
models=cells_linear_models,
stable_model=None,
key=key,
filename=f"E7-9h_midgut_{key}_linear_movie.mp4",
model_style="points",
model_size=5,
ambient=0.5,
colormap="default_cmap",
show_legend=True,
legend_kwargs=dict(title="", fmt="%.2e", legend_loc=(0.87, 0.3), label_font_size=20),
off_screen=True,
jupyter=False,
background="black",
cpo=cpo,
window_size=(1024, 1024),
)
Torsion¶
[16]:
key = "torsion"
glm_key = f"glm_degs_{key}"
st.tdr.morphofield_torsion(adata=stage_adata, vf_key="VecFld_morpho", key_added=key,)
st.tl.glm_degs(adata=stage_adata, layer=None, fullModelFormulaStr=f'~cr({key}, df=3)', key_added=glm_key, qval_threshold=0.01, llf_threshold=-2500)
glm_data = stage_adata.uns[glm_key]["glm_result"]
glm_dict[key] = glm_data
glm_data
|-----> [Calculating acceleration] in progress: 100.0000%
|-----> [Calculating acceleration] finished [0.0342s]
Calculating torsion: 100%|██████████| 2326/2326 [00:00<00:00, 123461.80it/s]
|-----? Gene expression matrix must be normalized by the size factor, please check if the input gene expression matrix is correct.If you don't have the size factor normalized gene expression matrix, please run `dynamo.pp.normalize_cell_expr_by_size_factors(skip_log = True)`.
|-----> [Detecting genes via Generalized Additive Models (GAMs)] in progress: 100.0000%
|-----> [Detecting genes via Generalized Additive Models (GAMs)] finished [332.7657s]
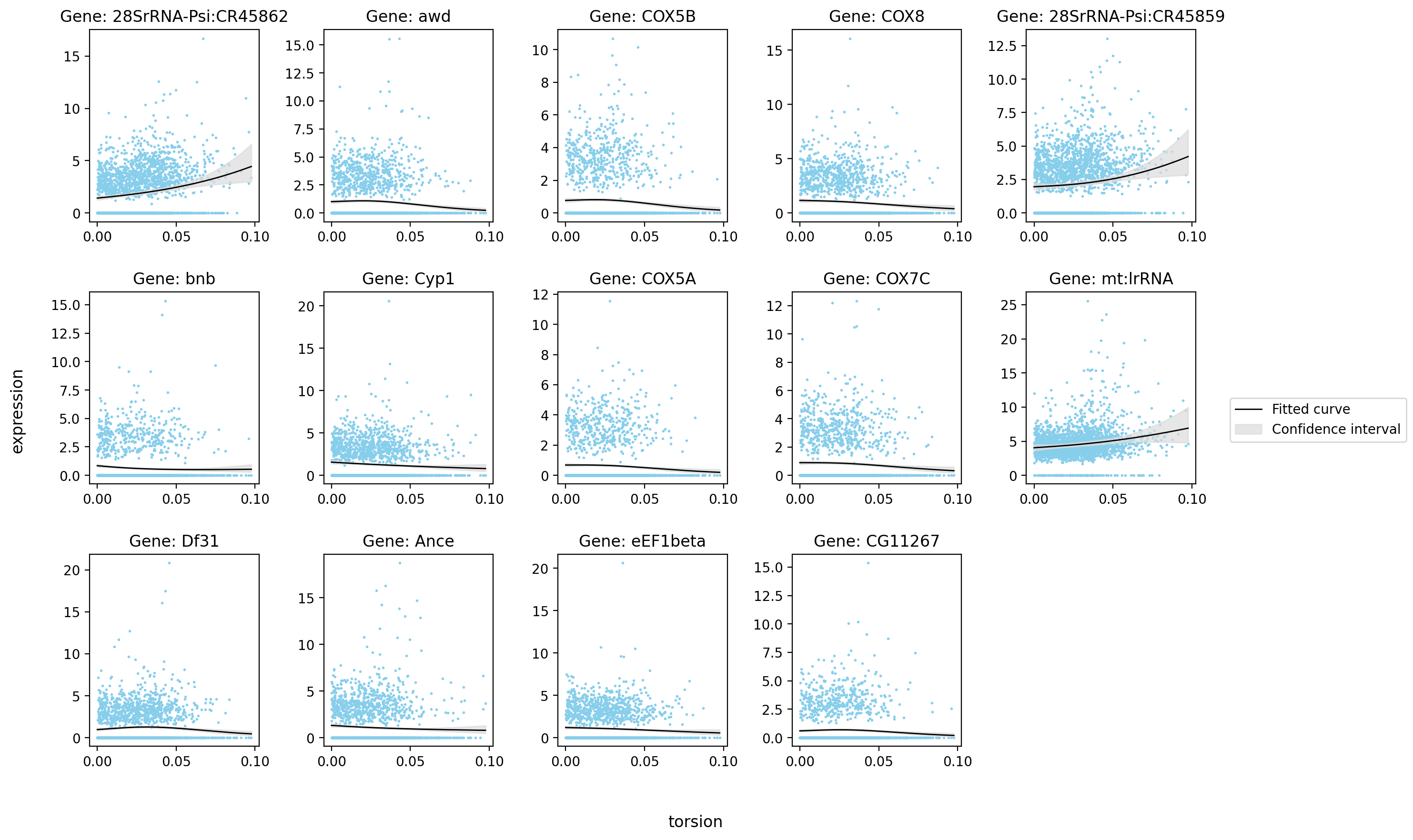
[16]:
| status | family | log-likelihood | pval | qval | |
|---|---|---|---|---|---|
| 28SrRNA-Psi:CR45862 | ok | NB2 | -4346.507812 | 3.861141e-15 | 2.472739e-12 |
| awd | ok | NB2 | -3186.442383 | 3.803082e-07 | 1.154048e-05 |
| COX5B | ok | NB2 | -2744.803711 | 2.241822e-06 | 4.618874e-05 |
| COX8 | ok | NB2 | -3231.869873 | 5.392043e-06 | 9.460696e-05 |
| 28SrRNA-Psi:CR45859 | ok | NB2 | -4634.142578 | 1.133787e-05 | 1.689693e-04 |
| bnb | ok | NB2 | -2515.703857 | 1.134524e-05 | 1.689693e-04 |
| Cyp1 | ok | NB2 | -3642.751221 | 5.894959e-05 | 6.097518e-04 |
| COX5A | ok | NB2 | -2502.031494 | 6.766557e-05 | 6.806412e-04 |
| COX7C | ok | NB2 | -2909.620850 | 2.804796e-04 | 2.050890e-03 |
| mt:lrRNA | ok | NB2 | -6082.588867 | 4.755917e-04 | 3.090644e-03 |
| Df31 | ok | NB2 | -3453.745605 | 5.343023e-04 | 3.363636e-03 |
| Ance | ok | NB2 | -3360.501221 | 5.547805e-04 | 3.470894e-03 |
| eEF1beta | ok | NB2 | -3307.284668 | 6.429467e-04 | 3.881419e-03 |
| CG11267 | ok | NB2 | -2541.744141 | 8.072325e-04 | 4.622639e-03 |
[17]:
st.pl.glm_fit(
adata=stage_adata,
gene=glm_data.index.tolist(),
ncols=5,
feature_x=key,
feature_y="expression",
feature_fit="mu",
glm_key=glm_key,
lowess=False,
frac=0.2,
save_show_or_return="both",
)
st.pl.torsion(
adata=stage_adata,
model=st.tdr.collect_models([stage_pc, trajectory_model]),
torsion_key=key,
colormap="default_cmap",
jupyter="static",
model_style=["points", "wireframe"],
model_size=[5, 2],
cpo=cpo,
window_size=(1024, 1024),
legend_kwargs=dict(title="", fmt="%.2e", legend_loc=(0.87, 0.3), label_font_size=20),
)
Saving figure to develop/develop_midgut/genesis_feature/E7-9h_midgut_torsion/E7-9h_midgut_glm_degs_fit_torsion.pdf...
Done

[18]:
st.tdr.add_model_labels(
model=trajectory_model,
key_added=key,
labels=np.asarray(stage_adata[np.asarray(trajectory_model.point_data["obs_index"])].obs[key]),
where="point_data",
inplace=True,
)
cells_linear_models = st.tdr.construct_genesis_X(
stages_X=[stage_adata.obsm["3d_align_spatial"], stage_adata.obsm["X_cells_mapping"]],
n_spacing=100-1,
key_added=key,
label=[np.asarray(stage_adata.obs[key])] * 100,
)
st.pl.three_d_animate(
models=cells_linear_models,
stable_model=trajectory_model,
key=key,
stable_kwargs=dict(
key=key,
model_style="wireframe",
model_size=2,
ambient=0.5,
opacity=0.3,
colormap="default_cmap",
show_legend=False,
),
filename=f"E7-9h_midgut_{key}_linear_movie_with_trajectory.mp4",
model_style="points",
model_size=5,
ambient=0.5,
colormap="default_cmap",
show_legend=True,
legend_kwargs=dict(title="", fmt="%.2e", legend_loc=(0.87, 0.3), label_font_size=20),
off_screen=True,
jupyter=False,
background="black",
cpo=cpo,
window_size=(1024, 1024),
)
st.pl.three_d_animate(
models=cells_linear_models,
stable_model=None,
key=key,
filename=f"E7-9h_midgut_{key}_linear_movie.mp4",
model_style="points",
model_size=5,
ambient=0.5,
colormap="default_cmap",
show_legend=True,
legend_kwargs=dict(title="", fmt="%.2e", legend_loc=(0.87, 0.3), label_font_size=20),
off_screen=True,
jupyter=False,
background="black",
cpo=cpo,
window_size=(1024, 1024),
)
Jacobian¶
[19]:
key = "jacobian"
st.tdr.morphofield_jacobian(adata=stage_adata, vf_key="VecFld_morpho", key_added=key,)
stage_adata.uns["jacobian"]
[19]:
array([[[ 2.08105242e-05, 2.70779418e-05, 3.14283350e-05, ...,
-8.25586263e-06, -8.49332510e-06, -5.65412876e-06],
[ 7.23267922e-05, 6.98323637e-05, 6.34322214e-05, ...,
3.69536432e-07, 2.37314756e-07, -9.78666713e-07],
[ 4.65323612e-05, 4.93631551e-05, 3.55578438e-05, ...,
-3.28196112e-06, -3.21871336e-06, -5.28409549e-06]],
[[ 1.67115495e-06, 7.37013386e-06, 1.69219829e-05, ...,
7.19850134e-06, 7.21621752e-06, 2.70224705e-06],
[ 5.42722509e-05, 5.09177864e-05, 5.47200113e-05, ...,
-7.52587956e-06, -8.50232404e-06, -6.79560366e-06],
[ 4.87511113e-05, 5.29128280e-05, 4.34329063e-05, ...,
9.67988261e-07, 3.44231301e-08, 3.61775236e-06]],
[[ 7.83754913e-06, 1.73498770e-05, 3.04055759e-05, ...,
7.92778250e-06, 6.45248208e-06, -1.02374499e-05],
[ 1.02038938e-04, 1.00946200e-04, 9.68161684e-05, ...,
1.84122214e-05, 1.03350391e-05, 1.36330631e-05],
[ 6.19007123e-05, 6.78370197e-05, 5.12851558e-05, ...,
4.45638671e-05, 4.71304287e-05, 5.08665136e-05]]])
[20]:
st.pl.jacobian(
adata=stage_adata,
model=st.tdr.collect_models([stage_pc, trajectory_model]),
jacobian_key="jacobian",
colormap="default_cmap",
jupyter="static",
model_style=["points", "wireframe"],
model_size=[2.5, 1],
cpo=[cpo],
window_size=(512*3, 512*3),
)

GO analysis¶
[21]:
gp.get_library_name(organism="fly")
[21]:
['Allele_LoF_Phenotypes_from_FlyBase_2017',
'Allele_Phenotypes_from_FlyBase_2017',
'Anatomy_AutoRIF',
'Anatomy_AutoRIF_Predicted_zscore',
'Anatomy_GeneRIF',
'Anatomy_GeneRIF_Predicted_zscore',
'Coexpression_Predicted_GO_Biological_Process_2018',
'Coexpression_Predicted_GO_Cellular_Component_2018',
'Coexpression_Predicted_GO_Molecular_Function_2018',
'GO_Biological_Process_2018',
'GO_Biological_Process_AutoRIF',
'GO_Biological_Process_AutoRIF_Predicted_zscore',
'GO_Biological_Process_GeneRIF',
'GO_Biological_Process_GeneRIF_Predicted_zscore',
'GO_Cellular_Component_2018',
'GO_Cellular_Component_AutoRIF',
'GO_Cellular_Component_AutoRIF_Predicted_zscore',
'GO_Cellular_Component_GeneRIF',
'GO_Cellular_Component_GeneRIF_Predicted_zscore',
'GO_Molecular_Function_2018',
'GO_Molecular_Function_AutoRIF',
'GO_Molecular_Function_AutoRIF_Predicted_zscore',
'GO_Molecular_Function_GeneRIF',
'GO_Molecular_Function_GeneRIF_Predicted_zscore',
'Human_Disease_from_FlyBase_2017',
'InterPro_Domains_2019',
'KEGG_2019',
'PPI_Network_Hubs_from_DroID_2017',
'Pfam_Domains_2019',
'Phenotype_AutoRIF',
'Phenotype_AutoRIF_Predicted_zscore',
'Phenotype_GeneRIF',
'Phenotype_GeneRIF_Predicted_zscore',
'Putative_Regulatory_miRNAs_from_DroID_2015',
'RNAi_Screens_from_GenomeRNAi_2017',
'TF2DNA_2018',
'Transcription_Factors_from_DroID_2015',
'WikiPathways_2018']
[22]:
for key, data in glm_dict.items():
if len(data.index.tolist()) > 0:
save_folder = f"E7-9h_midgut_{key}"
# GO-biologial process
go_bp = gp.enrichr(
gene_list=data.index.tolist(),
gene_sets="GO_Biological_Process_2018",
organism="fly",
outdir=save_folder,
no_plot=True,
verbose=True
)
# GO-cellular component
gp_cc = gp.enrichr(
gene_list=data.index.tolist(),
gene_sets="GO_Cellular_Component_2018",
organism="fly",
outdir=save_folder,
no_plot=True,
verbose=True
)
# GO-molecular function
go_mf = gp.enrichr(
gene_list=data.index.tolist(),
gene_sets="GO_Molecular_Function_2018",
organism="fly",
outdir=save_folder,
no_plot=True,
verbose=True
)
2022-10-16 14:11:43,473 Connecting to Enrichr Server to get latest library names
2022-10-16 14:11:44,859 Analysis name: , Enrichr Library: GO_Biological_Process_2018
2022-10-16 14:11:51,166 Save file of enrichment results: Job Id:231b5ebbe467f630d7a0ea9e7a9a39bd
2022-10-16 14:11:51,170 Done.
2022-10-16 14:11:51,171 Connecting to Enrichr Server to get latest library names
2022-10-16 14:11:52,551 Analysis name: , Enrichr Library: GO_Cellular_Component_2018
2022-10-16 14:11:56,542 Save file of enrichment results: Job Id:e5d4d3bb96e241ee70e016b34a31fc08
2022-10-16 14:11:56,546 Done.
2022-10-16 14:11:56,547 Connecting to Enrichr Server to get latest library names
2022-10-16 14:11:57,954 Analysis name: , Enrichr Library: GO_Molecular_Function_2018
2022-10-16 14:12:01,850 Save file of enrichment results: Job Id:44c1a53e9556e824aef5ea46234a3cd6
2022-10-16 14:12:01,852 Done.
2022-10-16 14:12:01,853 Connecting to Enrichr Server to get latest library names
2022-10-16 14:12:03,349 Analysis name: , Enrichr Library: GO_Biological_Process_2018
2022-10-16 14:12:07,927 Save file of enrichment results: Job Id:ed7c2d3f975ee261ffe541326810c97d
2022-10-16 14:12:07,932 Done.
2022-10-16 14:12:07,933 Connecting to Enrichr Server to get latest library names
2022-10-16 14:12:09,320 Analysis name: , Enrichr Library: GO_Cellular_Component_2018
2022-10-16 14:12:13,247 Save file of enrichment results: Job Id:027814f8da96a2305105a3fcc66febcb
2022-10-16 14:12:13,248 Done.
2022-10-16 14:12:13,249 Connecting to Enrichr Server to get latest library names
2022-10-16 14:12:14,632 Analysis name: , Enrichr Library: GO_Molecular_Function_2018
2022-10-16 14:12:18,544 Save file of enrichment results: Job Id:5fc10f71d4ca7b9f89ae182e57f3612f
2022-10-16 14:12:18,551 Done.
2022-10-16 14:12:18,553 Connecting to Enrichr Server to get latest library names
2022-10-16 14:12:19,954 Analysis name: , Enrichr Library: GO_Biological_Process_2018
2022-10-16 14:12:24,336 Save file of enrichment results: Job Id:f81cd3475f2e62680675ad00366355af
2022-10-16 14:12:24,342 Done.
2022-10-16 14:12:24,348 Connecting to Enrichr Server to get latest library names
2022-10-16 14:12:25,726 Analysis name: , Enrichr Library: GO_Cellular_Component_2018
2022-10-16 14:12:30,748 Save file of enrichment results: Job Id:89df4375762b1f95d12d56ee740c75a6
2022-10-16 14:12:30,752 Done.
2022-10-16 14:12:30,754 Connecting to Enrichr Server to get latest library names
2022-10-16 14:12:32,168 Analysis name: , Enrichr Library: GO_Molecular_Function_2018
2022-10-16 14:12:36,127 Save file of enrichment results: Job Id:2600b4c6db1bec44d1f67347d2392e99
2022-10-16 14:12:36,133 Done.
2022-10-16 14:12:36,135 Connecting to Enrichr Server to get latest library names
2022-10-16 14:12:37,611 Analysis name: , Enrichr Library: GO_Biological_Process_2018
2022-10-16 14:12:41,846 Save file of enrichment results: Job Id:c02d8a0119d766863cf00e35ca9e3794
2022-10-16 14:12:41,852 Done.
2022-10-16 14:12:41,854 Connecting to Enrichr Server to get latest library names
2022-10-16 14:12:43,244 Analysis name: , Enrichr Library: GO_Cellular_Component_2018
2022-10-16 14:12:47,747 Save file of enrichment results: Job Id:ee98c8b120087c155ce5116a5dfff9c7
2022-10-16 14:12:47,753 Done.
2022-10-16 14:12:47,757 Connecting to Enrichr Server to get latest library names
2022-10-16 14:12:49,105 Analysis name: , Enrichr Library: GO_Molecular_Function_2018
2022-10-16 14:12:53,686 Save file of enrichment results: Job Id:c4898910b80317f3cbee0ef3c690f369
2022-10-16 14:12:53,690 Done.
Genes expression pattern¶
[23]:
gene_list = list(set([gene for data in glm_dict.values() for gene in data.index.tolist()]))
[24]:
for gene in gene_list:
obs_index = stage_pc.point_data["obs_index"].tolist()
mkg_exp = stage_adata[obs_index, gene].X.flatten().astype(np.float64)
st.tdr.add_model_labels(model=stage_pc, labels=mkg_exp, key_added=gene, where="point_data", inplace=True)
mkg_amap = mkg_exp.copy()
mkg_amap = mkg_amap / mkg_amap.max()
mkg_amap[mkg_amap <= 0.5] = 0.5
rename_gene = str(gene).replace(":", "-") if ":" in gene else gene
st.pl.three_d_plot(
model=stage_pc,
key=gene,
model_style="points",
model_size=5,
# opacity=mkg_amap,
ambient=0.5,
colormap="default_cmap",
text=f"\nGene: {gene}",
show_legend=True,
legend_kwargs=dict(title=""),
off_screen=True,
jupyter=False,
background="black",
cpo=cpo,
window_size=(1024, 1024),
filename=f"pc_model_gene_{rename_gene}.pdf"
)
[ ]: